Commits on Source (5)
-
Johannes Wasmer authored
-
Johannes Wasmer authored
-
Johannes Wasmer authored
-
Johannes Wasmer authored
-

Showing
- bib/bibliography.bib 335 additions, 7 deletionsbib/bibliography.bib
- fig/external/papers/dominaJacobiLegendreFrameworkMachine2024/originals/fig4.1.pdf 3 additions, 0 deletions...naJacobiLegendreFrameworkMachine2024/originals/fig4.1.pdf
- fig/external/papers/dominaJacobiLegendreFrameworkMachine2024/originals/fig5.1.pdf 3 additions, 0 deletions...naJacobiLegendreFrameworkMachine2024/originals/fig5.1.pdf
- fig/external/papers/dominaJacobiLegendreFrameworkMachine2024/processed/fig-5.1-first-row-only.png 3 additions, 0 deletions...FrameworkMachine2024/processed/fig-5.1-first-row-only.png
- fig/external/papers/dominaJacobiLegendreFrameworkMachine2024/processed/fig5.1-with-polynomials.png 3 additions, 0 deletions...rameworkMachine2024/processed/fig5.1-with-polynomials.png
- fig/external/papers/dominaJacobiLegendreFrameworkMachine2024/processed/fig5.1-with-polynomials.xcf 0 additions, 0 deletions...rameworkMachine2024/processed/fig5.1-with-polynomials.xcf
- fig/logos/casus.science/Logo-CASUS-color.png 3 additions, 0 deletionsfig/logos/casus.science/Logo-CASUS-color.png
- fig/logos/hida/HiDA_Logo_RGB_kompakt.jpg 3 additions, 0 deletionsfig/logos/hida/HiDA_Logo_RGB_kompakt.jpg
- fig/logos/tcd.ie/Trinity-College-Dublin-Logo.png 3 additions, 0 deletionsfig/logos/tcd.ie/Trinity-College-Dublin-Logo.png
- fig/logos/tcd.ie/Trinity_Main_Logo.jpg 3 additions, 0 deletionsfig/logos/tcd.ie/Trinity_Main_Logo.jpg
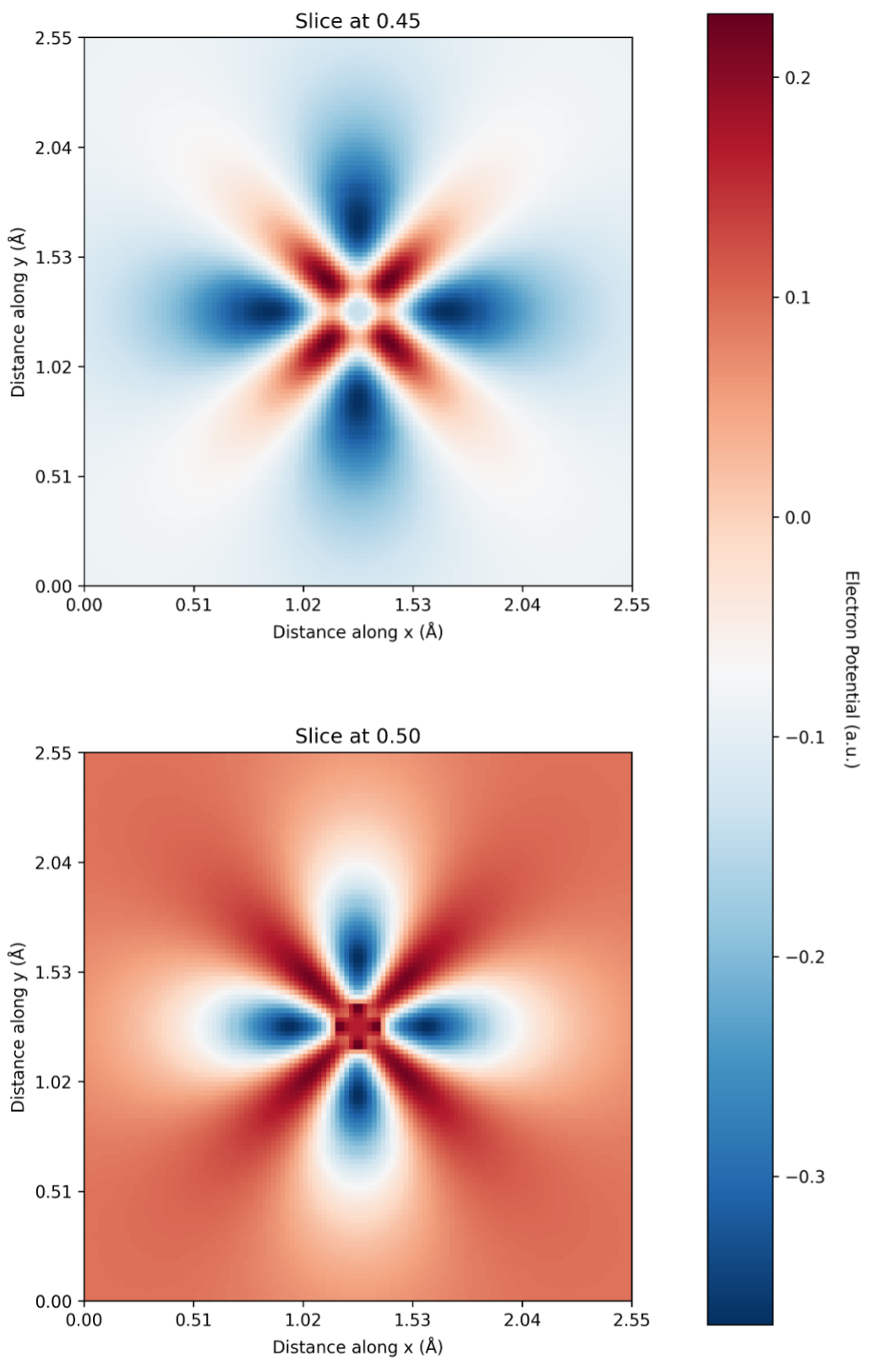
- fig/presentation-2024-09/tmp-vimp-prediction/kkr-cu-pot-column2.png 3 additions, 0 deletions...tation-2024-09/tmp-vimp-prediction/kkr-cu-pot-column2.png
- fig/presentation-2024-09/tmp-vimp-prediction/kkr-cu-pot-wrong-colorbar.png 3 additions, 0 deletions...2024-09/tmp-vimp-prediction/kkr-cu-pot-wrong-colorbar.png
File added
File added
{kind=link}

131 B
{kind=link}

131 B
File added
{kind=link}
130 B
{kind=link}
131 B
{kind=link}
130 B
{kind=link}
131 B
{kind=link}
131 B
{kind=link}

131 B